Elektroforeza
kapilarna (CE) jest stosunkowo nową, dynamicznie rozwijającą się techniką
rozdziału mieszanin chemicznych
Historycznie pierwszy raz elektroforeza została
zastosowana do rozdziału mieszaniny białek przez szwedzkiego uczonego
Tisseliusa, w 1930 roku. Po wypełnieniu rurki (rurka w kształcie litery U)
roztworem białek i umieszczeniu w jej obu końcach elektrod, połączonych z
zewnętrznym źródłem napięcia, stwierdził on iż białka wędrują w kierunku
elektrod z różnymi prędkościami.
W wolnym roztworze efekt rozdziału jest jednak
organiczny przez dyfuzję termiczną oraz konwekcję rozdzielanych cząsteczek. Z
tego powodu w dalszym rozwoju elektroforezy wprowadzono środowisko
antykonwekcyjne. Roztwór elektrolitu umieszczano w żelach, takich jak agaroza,
poliakrylamid czy bibule chromatograficznej.
Kolejnym etapem w rozwoju elektroforezy jest
zastosowanie w późnych latach 70 kapilar o małej średnicy (rzędu mikrometrów),
a technika ta nosi nazwę elektrofroforezy kapilarnej, łączy ona w sobie
szybkość, odtwarzalność oraz łatwość automatyzacji całego procesu.
Aparatura do elektroforezy kapilarnej jest stosunkowo prosta, schemat podstawowego zestawu przedstawiono na rysunku 1.

Rys.1. Schemat aparatury do elektroforezy kapilarnej.
Rozdział przeprowadza się w kapilarach ze stopionej krzemionki (kwarcu) SiO2 wypełnionych roztworem elektrolitu nośnego. Najczęściej stosowane są kapilary o średnicy wewnętrznej 25–75 µm i długości około 50 cm. Z zewnątrz, stopiona krzemionka pokryta jest warstwą poliamidową, celem zwiększenia jej wytrzymałości (Rys.2.).
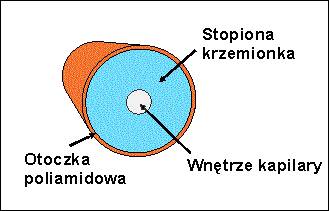
Rys.2. Przekrój poprzeczny kapilary ze stopionej krzemionki.
Końce kapilary umieszczone są w dwóch naczyniach, również
wypełnionych elektrolitem podstawowym. W jednym z naczyń znajduje się katoda
(-), a w drugim anoda (+). Elektrody (Pt) połączone są ze źródłem wysokiego
napięcia, w granicach od 10 do 30 kV, pracującym w zakresie prądowym od 0 do
200 mA.
Próbkę badaną wprowadza się do anodowego końca kapilary,
natomiast przy katodowym końcu najczęściej znajduje się detektor.
Cześć wysokonapięciowa układu, ze względów
bezpieczeństwa, umieszczana jest w obudowie z plexi (polimetakrylanu metylu),
często kapilara jest dodatkowo termostatowana.
Cały układ (detektor, zasilacz wysokiego napięcia,
system wstrzykiwania próbki oraz napełniania kapilary elektrolitem)
kontrolowany jest za pomocą komputera. W chwili obecnej na rynku dostępny jest
szeroki wybór komercyjnych, w pełni zautomatyzowanych, aparatów do
elektroforezy kapilarnej. Na rysunku 3 przedstawiono zdjęcie aparatu do
elektroforezy kapilarnej firmy Waters - Capillary Ion Analyzer.

Rys.3. Komercyjny aparat do CE - Waters Capillary Ion Analyzer.
Podstawą rozdziałów elektroforetycznych jest różnica w prędkościach migracji cząstek obdarzonych ładunkiem w stałym polu elektrycznym. Posiadające ładunek dodatni kationy przyciągane są przez katodę (-), a aniony przez anodę (+). Prędkość migracji v[cm·s-1] w kierunku elektrod jest różna dla różnych cząsteczek i zależy od natężenia pola elektrycznego E [V/cm] oraz ruchliwości elektroforetycznej:
v = µefE
Ruchliwość elektroforetyczna µef [m2·V-1·s-1] jest wielkością charakterystyczną dla danej cząsteczki i opisana jest następującym równaniem:
µef = q/(6·π·r·η)
z którego wynika, iż jest ona proporcjonalna do
wartości ładunku cząsteczki q [C], a odwrotnie proporcjonalna do
lepkości środowiska η [kg·m-1·s-1] i promienia cząsteczki r[cm] (czym mniejsza cząsteczka i
większy ładunek tym większa szybkość migracji).
W elektroforezie kapilarnej decydujące
znaczenie ma jeszcze jedno zjawisko, a mianowicie elektroosmoza. Gdy do końców kapilary wypełnionej roztworem
elektrolitu przyłożymy wysokie napięcie, obserwujemy przepływ elektroosmotyczny, który polega na przemieszczaniu się
całej masy elektrolitu w kierunku jednej z elektrod.
Przyczyną przepływu elektroosmotycznego jest
obecność podwójnej warstwy elektrycznej, która powstaje na granicy faz
roztwór/ścianka kapilary. Przy wartości pH większej od 3, wewnętrzna ścianka
kapilary ze stopionej krzemionki posiada ładunek ujemny, który związany jest z
dysocjacją grup silanolowych (SiOH), przy niższym pH jonizacja nie jest
całkowita.
Kationy elektrolitu podstawowego przyciągane są
przez ładunki ujemne na ściankach kapilary (SiO-), co powoduje
powstanie wewnętrznej (związanej) oraz zewnętrznej (dyfuzyjnej) warstwy
kationów, powstaje podwójna warstwa elektryczna (Rys.4.).
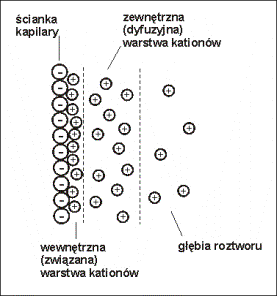
Rys.4. Schemat podwójnej warstwy elektrycznej powstającej przy ściance kapilary.
Kationy warstwy zewnętrznej przyciągane są przez
ujemną katodę, a ponieważ kationy te są solwatowane przez cząsteczki
rozpuszczalnika, „porywają” one razem ze sobą cała masę roztworu znajdującego
się w kapilarze.
Szybkość przepływu elektroosmotycznego jest
zazwyczaj większa od szybkości elektroforetycznych poszczególnych jonów, co
powoduje, że zarówno kationy, cząstki obojętne jak i aniony poruszają się w
kierunku katody (-).
W obecności przepływu elektroosmotycznego,
szybkość z jaką dany jon porusza się w kierunku katody vobs (szybkość
obserwowana) opisana jest następującą zależnością:
vobs = (µef + µeo)E [cm/s]
gdzie: µeo – ruchliwość elektroosmotyczna. W przypadku anionów µef jest wartością ujemna.
W rezultacie kolejność, w jakiej rozdzielane cząstki docierają do katodowego końca kapilary jest następująca: najpierw małe obdarzone dużym ładunkiem kationy, następnie większe kationy o mniejszym ładunku, nierozdzielone cząsteczki obojętne, duże aniony o małym ładunku, a na koniec małe aniony posiadające duży ładunek elektryczny (Rys.5.).
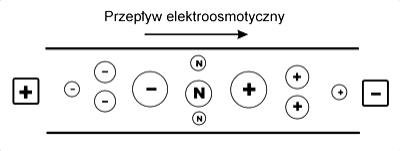
Rys.5. Kolejność w jakiej rozdzielane cząsteczki poruszają się w kierunku katody.
Dzięki temu, że przepływ elektroosmotyczny jest generowany przy ściankach kapilary na całej jej długości, charakteryzuje się on stała szybkością przepływu w całym jego przekroju (Rys.6).
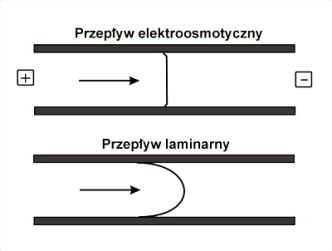
Rys.6. Przekrój podłużny przepływu elektroosmotycznego oraz laminarnego.
Płaski profil przepływu elektroosmotycznego jest
bardziej korzystny od przepływu laminarnego, spotykanego np. w technikach
chromatograficznych. W przypadku przepływu laminarnego, który jest wymuszony
przez zastosowanie ciśnienia na jednym z końców kapilary, roztwór znajdujący
się w jej środku porusza się szybciej niż roztwór będący bliżej ścianek.
Obecność przepływu laminarnego powoduje poszerzenie długości zajmowanej przez
próbkę w kapilarze i co za tym idzie również poszerzenie sygnałów z detektora.
Możliwe jest również odwrócenie
kierunku przepływu elektroosmotycznego przez odpowiednią modyfikację ładunku na
ściankach kapilary, np. przez dodanie do roztworu elektrolitu surfaktanata
kationowego (związek powierzchniowo czynny). Surfaktant ulega adsorpcji na
ściankach kapilary, przez co ścianki zyskują ładunek dodatni. Przepływ
elektroosmotyczny może być także wyeliminowany przez odpowiednią modyfikacje
ścianek kapilary.
Głównym ograniczeniem w szybkości,
rozdzielczości oraz skali rozdziałów elektroforetycznych jest ciepło Joule’a
generowane, gdy przez roztwór elektrolitu przepływa prąd elektryczny. W
przypadku stosowanych w CE kapilar, wytwarzane w roztworze ciepło z łatwością
rozpraszane jest do otoczenia. Możliwe jest to dzięki korzystnemu stosunkowi
powierzchni kapilary do jej objętości przy tak małych średnicach wewnętrznych.
Dodatkowym czynnikiem wpływającym korzystnie na
rozdzielczość oraz zmniejszenie wysokości półki teoretycznej jest brak fazy
stacjonarnej. Elektroforeza kapilarna w rutynowych zastosowaniach pozwala
osiągnąć liczbę półek teoretycznych w granicach od 50000 do 500000.
Roztwór próbki badanej w większości
przypadków wprowadzany jest do anodowego końca kapilary. Typowe objętości
wstrzykiwanej próbki wahają się od 10 do 100 nl. Najczęściej stosowaną metodą
jest umieszczenie anodowego końca kapilary w naczyniu z roztworem próbki,
znajdującym się wyżej względem katodowego końca kapilary, metoda
hydrodynamiczna – grawitacyjna (trzy podstawowe metody wprowadzania próbki
przedstawiono schematycznie na rysunku 7).
Inna hydrodynamiczna metoda wstrzykiwania polega
na umieszczeniu końca kapilary w szczelnym zbiorniku z próbką i zastosowaniu
wysokiego ciśnienia.
Popularna jest także metoda elektrokinetyczna, w
przypadku której, w naczyniu z próbka umieszczana jest anoda, a następnie do
końców kapilary przykładane napięcie rzędu 5 kV, mieszanina rozdzielana
wprowadzana jest do kapilary dzięki przepływowi elektroosmotycznemu.
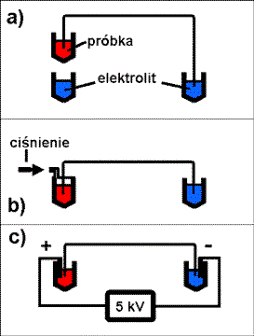
Rys.7. Metody wprowadzania próbki: a) hydrodynamiczna – grawitacyjna, b) hydrodynamiczna – ciśnieniowa, c) elektrokinetyczna.
Dzięki
temu, ze w CE wszystkie składniki rozdzielanej mieszaniny przechodzą przez
jeden wspólny punkt (katodowy koniec kapilary), większość stosowanych
detektorów jest zbliżona do tych używanych w HPLC.
W tabeli 1 zestawione są najczęściej spotykane w
elektroforezie kapilarnej techniki detekcji oraz odpowiadające im limity
detekcji.
Tabela 1. Podstawowe metody detekcji w CE [6].
| Metoda detekcji | Limit detekcji (mol/dm3) |
| Spektrofotometria | |
| Absorbcyjna | 10-5 – 10-6 |
| Fluorescencyjna | 10-10 – 10-16 |
| NMR | 10-3 |
| Elektrochemia | |
| Konduktometria | 10-7 – 10-8 |
| Potencjometria | 10-7 – 10-8 |
| Amperometria | 10-7 – 10-8 |
| Radiometria | 10-10 |
Elektroforeza kapilarna w wolnym roztworze. Jest to podstawowa odmiana, w której rozdział polega na różnicy w szybkościach migracji poszczególnych jonów, a zasady jej działania zostały omówione powyżej. Głównym ograniczeniem jest możliwość rozdziału jedynie cząstek posiadających ładunek elektryczny. Elektroforogram otrzymany przez elektroforetyczny rozdział 27 kationów zamieszczono na rysunku 8.
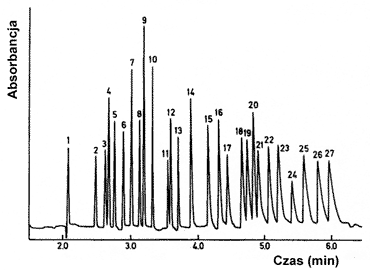
Rys.8. Elektroforogram otrzymany dla mieszaniny 27 kationów (detekcja spektrofotometryczna, długość fali 214 nm). Kolejne sygnały odpowiadają następującym kationom: (1) K+, (2) Ba2+, (3) Sr2+, (4) Na+, (5) Ca2+, (6) Mg2+, (7) Mn2+, (8) Cd2+, (9) Li+, (10) Co2+, (11) Pb2+, (12) Ni2+, (13) Zn2+, (14) La3+, (15) Ce3+, (16) Pr3+, (17) Nd3+, (18) Sm3+, (19) Gd3+, (20) Cu2+, (21) Tb3+, (22) Dy3+, Ho3+, (24) Er3+, (25) Tm3+, (26) Yb3+, (27) Lu3+.[7]
Micelarna elektrokinetyczna chromatografia
kapilarna jest odmianą CE
pozwalająca na rozdział cząstek obojętnych. Stosowane są jonowe związki
powierzchniowo czynne w stężeniu przekraczającym krytyczne stężenie micelarne
(CMC).
Do najczęściej stosowanych surfaktantów anionowych
należy SDS (C12H25OSO3-Na+).
Budowę pojedynczej cząsteczki surfaktanta oraz
micele tworzące się powyżej CMC przedstawiono schematycznie na rysunku 9.
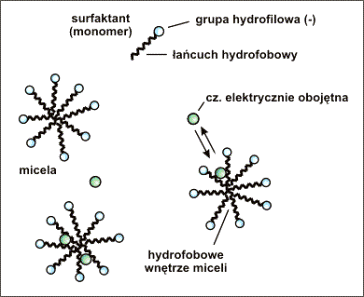
Rys.9. Monomer surfaktanta anionowego, micele tworzące się powyżej CMC oraz równowaga jaka ustala się gdy w roztworze znajduje się substancja elektrycznie obojętna. W przypadku surfaktanta anionowego takiego jak SDS micele posiadają sumaryczny ładunek ujemny.
Gdy w roztworze brak jest surfaktanta, lub jego
stężenie nie jest wystarczające aby powstały micele, cząstki obojętne poruszają
się z szybkością równą szybkości przepływu elektroosmotycznego.
Natomiast w przypadku obecności miceli, rozdzielana
substancja elektrycznie obojętna ulega podziałowi między roztwór elektrolitu, a
niepolarne wnętrze miceli (Rys.9.).
Cząsteczka obojętna znajdując się we wnętrzu miceli
przemieszcza się razem z nią, a rozdział dokonuje się na podstawie różnic
współczynników podziału dla różnych substancji.
Przy najczęściej stosowanym surfaktancie anionowym (SDS)
micele posiadają ładunek ujemny. Cząsteczka niepolarna, znajdująca się we
wnętrzu takiej miceli porusza się wolniej od przepływu elektroosmotycznego
(przepływ elektroosmotyczny w kierunku katody).
Kapilarna
elektroforeza żelowa. Jest to
odmiana elektroforezy żelowej, przez ostatnie trzy dekady stosowanej szeroko w
biochemii. W technice tej wypełnienie kapilary stanowi żel (najczęściej liniowy
polimer), a o rozdziale substancji decydują rozmiary cząsteczek – sączenie
molekularne.
Metoda ta znalazła zastosowanie w rozdziale
makromolekuł takich jak peptydy, białka oraz DNA.
Izoelektryczne ogniskowanie. Wariant ten stosowany jest do rozdziału
cząsteczek amfiprotycznych, zdolnych posiadać zarówno ładunki dodatnie, jak i
ujemne (np.: aminokwasy, peptydy, białka). Przy pewnej charakterystycznej
wartości pH, ładunek dodatni takich cząsteczek jest dokładnie równy ładunkowi
ujemnemu, cząsteczka jest elektrycznie obojętna i nie porusza się w polu
elektrycznym.
W przypadku ogniskowania izoelektrycznego kapilara
wypełniona jest roztworem buforowym o różnej wartości pH, której gradient we
wnętrzu kapilary pozostaje stały w czasie, gdy przykładane jest napięcie.
Próbka wprowadzana jest do końca kapilary o niższej wartości pH, a składniki
rozdzielanej mieszaniny posiadające ładunek dodatni wędrują w kierunku
detektora.
W momencie, gdy składniki mieszaniny rozdzielanej
znajdą się w części kapilary gdzie wartość pH odpowiada punktowi
izoelektrycznemu (ilość ładunków dodatnich jest dokładnie równa ilości ładunków
ujemnych) danej cząstki, wędrówka ustaje. Następnie odłączone zostaje wysokie
napięcie, a roztwór wypchnięty z kapilary przez zastosowanie wysokiego
ciśnienie na jednym z jej końców.
CE – (ang. Capillary electrophoresis) elektroforeza kapilarna
elektrolit – Terminu elektrolit używa się w dwóch nieco
innych znaczeniach. W pierwszym, oznacza on substancję, która rozpuszczona
dysocjuje, dając roztwór przewodzący jonowo. Rozróżnia się elektrolity słabe i
mocne.
W drugim znaczeniu elektrolitem nazywa się każdy
ośrodek przewodzący jonowo (roztwór zawierający jony lub stopiona sól). W tym
znaczeniu mówi się o elektrolicie akumulatora lub elektrolicie w przestrzeni
elektrodowej elektrolizera.
podwójna warstwa elektryczna – Składa się ona z dwóch warstw ładunków o znakach przeciwnych. Jedna z tych warstw może być rozmyta, jak np. w modelu cząstek koloidalnych, w którym ładunek cząstki przyciąga rozmytą atmosferę jonową w roztworze otaczającym cząstkę. Do opisu podwójnej warstwy elektrycznej zaproponowano kilka modeli (model Helmholtza, model Gouya-Chapmana i model Sterna).
HPLC – (ang. high performance liquid chromatography) wysokosprawna chromatografia cieczowa
krytyczne stężenie micelarne (CMC) – Jest to stężenie roztworu cząsteczek mających na jednym końcu grupę hydrofobową, a na drugim hydrofilową, powyżej którego tworzą się w nim micele. Micele są klasterami cząsteczek o wymiarach typowych dla cząstek koloidalnych.
półka teoretyczna – Liczba półek teoretycznych jest miarą skuteczności technik rozdziału (takich jak destylacja frakcjonowana lub chromatografia). Liczba półek teoretycznych wyraża liczbę kroków podziału między dwie fazy potrzebnych, aby z roztworu o danym składzie wyjściowym otrzymać roztwór o zadanym składzie.
substancje amfiprotyczne – Jest to cząsteczka zdolna zarówno do przyjęcia jak i oddania protonu. Typowy aminokwas jak glicyna jest substancja amfiprotyczną. W wodnym roztworze glicyny ustalają się trzy następujące równowagi:
NH2CH2COOH
↔ NH3+CH2COO-
NH3+CH2COO-
+ H2O ↔ NH2CH2COO- + H3O+
NH3+CH2COO-
+ H2O ↔ NH3+CH2COOH + OH-
surfaktant – substancja powierzchniowo czynna (np. mydło, detergenty syntetyczne). Cząsteczka surfaktanta może się składać z fragmentów polarnych i niepolarnych. W niskich stężeniach jest równomiernie rozmieszczona w roztworze. W wysokich tworzy micele.
współczynnik podziału – Jest to fundamentalna wielkość leżąca u podstaw technik chromatograficznych. Współczynnik podziału można wyrazić równaniem Nernsta: K = Cs/Cm, w którym Cs i Cm oznaczają stężenie danej substancji w fazie stacjonarnej (hydrofobowe wnętrze miceli – faza pseudostacjonarna) i ruchomej (elektrolit podstawowy).
[1] Peter William Atkins, Przewodnik po chemii fizycznej, PWN 1997
[2] Daniel C. Harris, Quantitative Chemical Analysis, W.H Freeman and Company, NY 1999
[3] Walenty Szczepaniak, Metody instrumentalne w analizie chemicznej, PWN 1996
[4] Paul D. Grossman, Joel C. Colburn, Capillary Electrophoresis – Theory and
Practice, Academic Press INC., 1992
[5] F.Foret, L. Krivankova, P. Bocek, Capillary Zone Electrophoresis, VCH
Publishers, Inc., NY1993
[6] Kelly Swinney, Darryl J. Bornhop, Detection in
capillary electrophoresis (Review), Electrophoresis 2000, 21, 1239-1250
[7] Miroslav Macka, Paul R. Haddad, Determination
of metal ions by capillary electrophoresis (Review), Electrophoresis 1997, 18,
2482-2501
Piotr Diakowski diakowski@home.com
Praca wpłynęła do ChemFana: 18-06-2001
