gdzie HA jest niezdysocjowaną cząsteczką kwasu, H+ to decydujący o kwasowości środowiska jon wodorowy, zaś A- to anion reszty kwasowej. (Dla uproszczenia wszystkie podawane tutaj równania będą dotyczyć kwasów jednoprotonowych)
| Kd= | [H+] [A] [HA] | (2) |
gdzie [H+], [A] i [HA] to stężenia odpowiednich form. Jednostką Kd jest mol/dm3, o ile w takich właśnie jednostkach wyrażamy stężenia. Stałe dysocjacji są rzeczywiście stałe - zachowują swoją wartość pomimo zmian stężenia kwasu (jest tu pewne "ale" - dotyczy to tylko roztworów rozcieńczonych, lecz musimy od czegoś zacząć). Teraz wiemy już, jak obiektywnie porównywać moc kwasów: należy zestawiać ze sobą odpwiednie stałe dysocjacji. Im stała dysocjacji większa, tym kwas mocniejszy. W tabeli 1 zebrane są stałe dysocjacji kilku kwasów.
Tabela 1
|
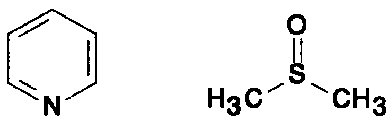 | |
| pirydyna | dmso |
Tabela 2
pierwszego protonu | ||||||||||||||||||||||
gdzie log oznacza logarytm dziesiętny zaś aH+ jest rozważaną aktywnością jonu wodorowego (wyrażoną w mol/dm3, ale jednostek nie logarytmujemy!). Większość początkujących chemików słyszała, że pH bardzo czystej wody równe jest 7 w temperaturze 25oC (aktywność jon~w wodorowych równa 10-7 mol/dm3), a roztworów kwasów i zasad odpowiednio mniejsza i większa od tej wartości. Zapewne spotkała się też ze sformułowaniem, że pH może się zmieniać od 0 do 14.
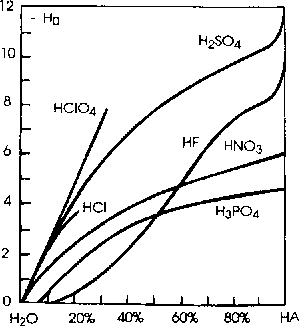
Zależność - H0 od ułamka molowego kwasu w roztworze |
HSO3F jest bezbarwną cieczą, wrzącą w temperaturze 163oC, dobrze rozpuszczającą się w wodzie i dającą z zasadami trwałe sole - fluorosiarczany. 100% HSO3F (H0=-15,1) jest kwaśniejszy i od HF (H0= -11,0) i od H2SO4 (H0=-11,9), ale są jeszcze inne możliwości otrzymania jeszcze mocniejszych kwasów - tak zwanych superkwasów (superkwasami przyjęto nazywać układy kwaśniejsze od 100% kwasu siarkowego).
Co zatem trzeba zrobić, by w układzie przybyło jonów H+? Trzeba związać jony F- układ będzie się starał przywrócić zachwianą równowagę, wytwarzając "przy okazji" jony wodorowe. Teraz trzeba wybrać substancję wiążącą jony F- - okazuje się, że doskonałe skutki dają niektóre fluorki, np. boru, niobu i arsenu:
Tabela 3. Funkcja kwasowości H0
|
SbF5 + F- --> SbF6-
Mieszaninę HF i SbF5 charekteryzuje H0 zbliżone do -22. To nie koniec możliwości! Korzystając z SbF5 można otrzymać jeszcze kwaśniejsze środowisko, ale trzeba zamiast fluorowodoru wziąć wspomniany wyżej kwas fluorosiarkowy. Mieszanina HSO3F i SbF5 (bezbarwna, dość ruchliwa ciecz) ze względu na swoje właściwości bywa nazywana "kwasem magicznym". Kwasowość tego układu jest tak duża, że przez długi czas nie umiano jej zmierzyć! Sądzono, że największa powinna być po zmieszaniu równomolowych ilości kwasu fluorosiarkowego i pięciofluorku antymonu, ale w 1981r okazało się inaczej. Znacznie większą aktywność jonów wodorowych wykazują mieszaniny zawierające duży nadmiar SbF5. Roztwór zawierający 9 cząsteczek SbF5 na 1 cząsteczkę HSO3F (ok. 200g SbF5 na 10g H HSO3F) jest 1015 razy kwaśniejszy od 100% kwasu siarkowego - tyle samo razy, ile ten ostatni od wody sodowej!
RH + H+ --> RH2+
Witold Mizerski KCh 3/92
