Piotr Solarz
|
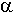 i
i 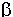 anomerów.
O ile dwupeptyd może istnieć w dwóch formach (pomijając istnienie
D-aminokwasów), o tyle dwie identyczne heksozy mogą się ze sobą łączyć na 11
różnych sposobów (Paulus i Klockow 1996). Fakt ten obrazuje tabela 1.
Dodatkowo grupy cukrowe mogą być modyfikowane przez acylację, metylację,
fosforylację, sulfonizację, jak i aminację (Paulus i Klockow 1996).
anomerów.
O ile dwupeptyd może istnieć w dwóch formach (pomijając istnienie
D-aminokwasów), o tyle dwie identyczne heksozy mogą się ze sobą łączyć na 11
różnych sposobów (Paulus i Klockow 1996). Fakt ten obrazuje tabela 1.
Dodatkowo grupy cukrowe mogą być modyfikowane przez acylację, metylację,
fosforylację, sulfonizację, jak i aminację (Paulus i Klockow 1996).
Tabela 1*
* Kobata 1994 ** Liczba rodzajów monocukrów w oligosacharydzie. | ||||||||||||||||||||||||||||||||
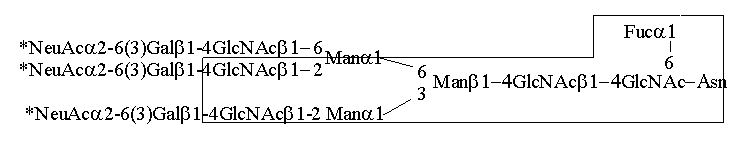
1-6(Man1-3)Man1-4GlcNAc1-4GlcNAcAsn.
Asparagina ta pochodzi z fragmentu białka o ściśle konserwatywnej sekwencji
(Asn-X-Ser/Thr), gdzie X może być dowolnym aminokwasem za wyjątkiem proliny i
hydroksyproliny. Pomimo wspólnej sekwencji rdzeniowej N-glikany odznaczają się
dużą zmiennością oligosacharydów przyłączonych do rdzenia. Dobrze poznane
zostały co najmniej cztery typy N-glikanów:
| Man1-2Man1- | 6 3 | Man1 | ||
| Man1-2Man1- |    | 6 3 | Man1-4GlcNAc1-4GlcNAc-Asn | |
| Man1-2Man1-2Man1- | 2 | Man1 | ||
| GlcNAc1 | Fuc1 | ||||||
|
Neu-NAc2-6Gal1-4GlcNAc1-6 Neu-NAc 2-6Gal1-4GlcNAc1-4Neu-NAc 2-6Gal1-4GlcNAc1-2
| Man1 |
|
6 3 |
| 4 | | 6 | ||
| Man1 | -4GlcNAc1 | -4GlcNAc | -Asn | ||||
|
Neu-NAc2-3Gal1-4GlcNAc1-4 Neu-NAc 2-6Gal1-4GlcNAc1-2
| Man1 |  | |||||
|
Man1-6 Man 1-3 | Man1 |
 |
6 3 |
| |||
| Man1 | -4GlcNAc1 | -4GlcNAc | -Asn | ||||
|
Neu-NAc2-3Gal1-4GlcNAc1-4 GlcNAc 1-2 | Man1 | 4 | | 6 | | ||||
| GlcNAc1 | Fuc1 | ||||||
1-4GlcNAc1-) przyłączoną do rdzenia.
Powtarzającą się sekwencja może być rozgałęziana za pomocą
GlcNAc-trasferazy Gal1-4GlcNAc1-6(Gal1-4GlcNAc1-3)Gal.
1-4.
Oprócz tego wszystkie typy N-glikanów mogą zawierać przyłączoną fukozę do
rdzeniowej N-acetyloglukozaminy (wiązaniem 1-6 w większości N-glikanów
zwierzęcych lub wiązaniem 1-3 w glikoproteinach roślin i niektórych
bezkręgowców).N-glikany pochodzące z roślinnych glikoprotein należą do dwóch
grup: wysokomannozowych i złożonych. Czynnikiem odróżniającym je od
glikoprotein zwierzęcych jest możliwość przyłączenia Xyl1-2 oraz brak
kwasów sjalowych (Leurage i Faye 1996). Grzyby posiadają aparat enzymatyczny
zdolny do wytwarzania tylko reszt wysokomannozowych.
-Ser/Thr (Schacter 1994) lub związaną
wiązaniem glikozydowym Fuc (Harris i Spellman 1993)
2-6GalNAc-Ser/Thr
1-3GalNAc-R - najbardziej popularny (Schacter 1986)
1-6(Gal1-3)GalNAc-R
1-3GalNAc-R
1-6(GlcNAc1-3)GalNAc-R
1-3GalNAc-R
1-6GalNAc-R / gdzie R to -Ser/Thr.1-3 lub 1-4, albo GlcNAc przyłączanej wiązaniem
1-3, lub 1-6. Koniec nieredukujący może być formowany poprzez
dodanie D-Gal, GalNAc, L-Fuc (Schacter 1994).1-2Man1-6Man1-4GlcNH21-6mio-inozytol-1-PO4-lipid,
w której fragmentem zmiennym w zależności od rodzaju tkanki jest część
lipidowa. Synteza kotwic GPI zapoczątkowywana jest w szorstkim retikulum
endoplazmatycznym. Proces ten wymaga szeregu przeniesień monocukrów na
fosfadydyloinozytol oraz przeniesienia fosforanu etanoloaminy z
fosfatydyloetanoloaminy na końcową mannozę.
1-2 do pierwszej mannozy
rdzenia, jak również sposób wiązania fukozy (wiązaniem 1-3) do pierwszej
GlcNAc na końcu redukującym (Ogawa i wsp. 1996). Inną różnicą jest całkowity
brak kwasów sjalowych.
 =490 nm i porównując ją z krzywą
wzorcową otrzymaną dla roztworów glukozy. Metoda ta umożliwia również
ilościowe oznaczanie zawartości cukrów w białkach (schemat zamieszczono
poniżej).
=490 nm i porównując ją z krzywą
wzorcową otrzymaną dla roztworów glukozy. Metoda ta umożliwia również
ilościowe oznaczanie zawartości cukrów w białkach (schemat zamieszczono
poniżej).
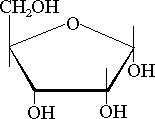 | -3H2O | 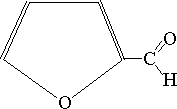 |
| -D-ryboza | Furfural |
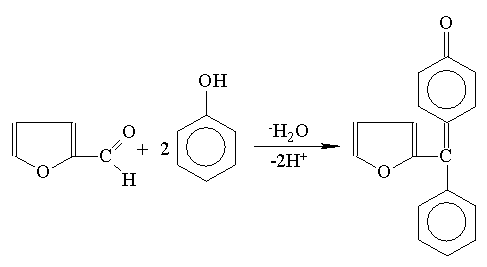
Istnieją odmiany metody fenolowej jak:
-naftolem: dająca reakcję z każdym cukrem
  |
HIO4 |
 |
F |
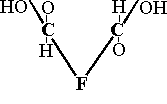 |
Tabela 2. (Paulus i Klockow 1996)
| Znacznik | Struktura | Długość fali [nm] | Granica wykrywalności [M] |
| 2-aminopirydyna (2-AP) | 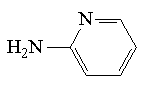 | 240 | 8*10-6 |
| kwas p-aminobenzowy | 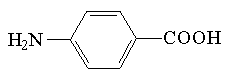 | 285 | 4*10-6 |
| ester etylowy kwasu p-aminobenzowego | 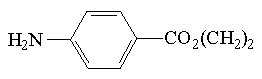 | 305 | 2*10-6 |
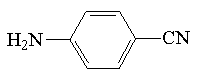
| p-aminobenzonitryl |
| 285 | 3*10-7 |
| kwas 8-aminonaftalen- 1,3,6-trisulfonowy (ANTS) | 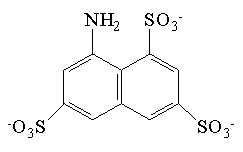 | 223 | 5*10-7 |
Oprócz wyżej wymienionych znaczników do znakowania UV używa się takich substancji jak:
max = 245 nm = 200 nmmax = 247 nm = 235 nm = 235nm (El Rassi i Mechref 1996)Tabela 3. (Paulus i Klockow 1996)
| Znacznik | Struktura |
| Długość fali wzbudzenia i emisji [nm] | Granica wykrywalności [M] |
| kwas 8-aminonaftalen-1,3,6-trisulfonowy (ANTS) | |
| (laser He-Cd) 325/520 (laser Ar) 275/520 | 5*10-81*10-9 |
| 3-(4-carboksybenzoilo)-2-chinolino carboksy aldehyd (CBQCA) | 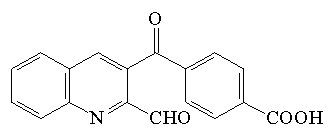 |
| (laser Ar) 457/552 | 3*10-9 |
| ester sukcynimidylowy 5-karboksytetra-metylorodaminy (TRSE) | 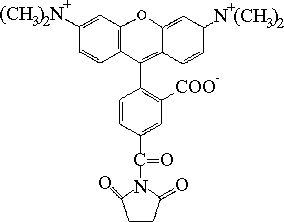 |
| (laser He-Ne) 543/580 | 5*10-11 |
Oprócz wymienionych w tabeli markerów do znakowania fluorescencyjnego używa się innych związków:
wzb = 425 nm; em = 520 nmwzb = 325 nm; em = 375 nmwzb = 325 nm; em > 495 nmwzb = 325 nm; em = 475 nmwzb = 315 nm; em = 420 nmwzb = 325 nm; em = 475 nmwzb = 455 nm; em = 512 nm (El Rassi i Mechref 1996)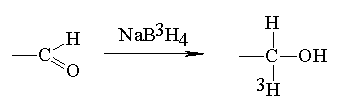 -eliminacja), czy hydrazynoliza
(która obecnie cieszy się znowu uznaniem). W ostatnich dwóch dekadach
"do głosu doszły" metody enzymatyczne, wykorzystujące glikozydazy.
W analizie N-glikanów wykorzystujemy endoglikozydazy mające specyficzne
działanie (rysunek poniżej).
-eliminacja), czy hydrazynoliza
(która obecnie cieszy się znowu uznaniem). W ostatnich dwóch dekadach
"do głosu doszły" metody enzymatyczne, wykorzystujące glikozydazy.
W analizie N-glikanów wykorzystujemy endoglikozydazy mające specyficzne
działanie (rysunek poniżej).
| X1 | 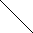  | PN Gaza | ||||||
| X2 | Man1 |  | Z | | 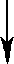 | ||||
| Y | Man1-4GlcNAc1 | - | 4GlcNAc | - | Asn | |||
| X3 | 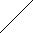 | Man1 | 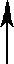 | |||||
| X4 | endoglikozydaza | |||||||
1-3 posiada wolną grupę hydroksylową
przy węglu C-2 (Maramatsu 1988). Miejsce cięcia tego enzymu, jak i innych
omawianych tutaj endoglikozydaz, znajduje się za pierwszą GlcNAc rdzenia.
1-3. Optymalne pH dla działania tego enzymu wynosi około 8.6.
1-3GalNAc
(Schacter 1986), może być zhydrolizowana przez enzymy prokariotyczne
(endo-GalNAc-aza D z Diplococus (Straptococcus) pneumoniae (Endo i
Kobata 1976) jak i eukariotyczne (endo-GalNAc-aza A z Alcaligens (Fan,
Kadowaki i wsp. 1988). Powyższe enzymy są w stanie hydrolizować tylko
dwucukrową strukturę Gal1-3GalNAc bez żadnych podstawień (O"Neill 1996).
O-glikany są jednak często bardziej zróżnicowane, a ich fragment
Gal1-3GalNAc rozbudowany. Optymalne pH dla tego enzymu wynosi 5.5.
Ostatnio pojawiły się doniesienia o nowym, słabo zdefiniowanym enzymie ze
Streptomyces opisanym jako endo-GalNAc-aza S (Iwase i wsp. 1988), który
jest w stanie hydrolizować większe struktury, niż opisane powyżej, na przykład
Gal1-3(Gal-1-4GlcNAc1-6)-GalNAc.
/)
-mannozydazy z Conavalia ensiformis hydrolizie
ulegają wiązania Man1<--2,3,6.
W innym przypadku niektóre wiązania mogą nie ulegać hydrolizie (Dwek i wsp.
1993). Czynnikami, których należy ściśle przestrzegać są: czystość enzymu,
stężenie substratu, pH, siła jonowa, rodzaj użytego buforu, temperatura, czas
reakcji. Niezachowanie któregoś z tych warunków może uczynić eksperyment
niepowtarzalnym i spowodować błędy w określaniu struktury analizowanej próby.
Schemat sekwencjonowania metodą RAAM
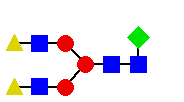
(na podstawie katalogu Oxford GlycoSystems 1994) dla N-glikanu
o przykładowej strukturze:
 |
 |
mannoza; n GlcNAc; u fukoza; s galaktoza.
obecność enzymu o danej specyfice; m - nieobecność danego enzymu
-Nacetyloheksozoamidaza (S. pneumoniae)
-3/4-fukozydaza (ziarno migdałów)
-fukozydaza (jądra wołu)
-Nacetyloheksozoamidaza (kanawalia mieczokształtna)
Tabela 4. Przykłady glikozydaz używanych w analizie glikanów (Dwek i wsp. 1993).
| Enzym | Oznaczenie EC | Źródło | Specyficzność |
| -D-sjalidaza | 3.2.1.18 | Arthobacter ureafaciens Clostridium perfringens Vibrio cholerae wirus Choroby Newcastle | NeuNAc/NeuGc |
| -D-galaktozydaza | 3.2.1.23 | jądra wołu Escherichia coli(Conavalia ensiformis) Streptococcus pneumoniae | Gal |
| -D-galaktozydaza | 3.2.1.22 | ziarno kawy | Gal1-->3, 4, 6 |
| -N-acetylo- -D-heksoaminidaza | 3.2.1.30 | Canavalia enciformis Streptococcus pneumoniae | Glc(Gal)NAc |
| -N-acetylo-D-heksoaminidaza | 3.2.1.49 | wątroba kurczaka | GalNAc |
| -D-mannozydaza | 3.2.1.24 | Canavalia enciformis Asparagillus phoenicins I | Man1-->2, 3, 6 Man 1-->2 |
| -D-mannozydaza | 3.2.1.25 | Helix pomatia Achatina fulica | Man1-->4 |
| -L-fukozydaza | 3.2.1.51 | Charonia lampas nadjądrza wołu migdały I migdały II | Fuc |
| -D-ksylozydaza | Charonia lampas | Xyl |
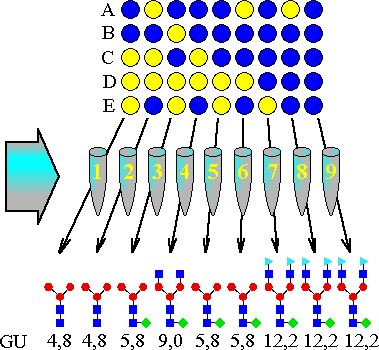
Tabela 5. Struktura oligosacharydów 1--12 i ich zachowanie w lektynowej
seryjnej chromatografii powinowactwa; rozmiar kolumny:
0.7cm * 2.6cm. (Glycobiology 1993 s. 316, edy. Minoru Fokuda i Akira Kobata,
Oxford University Press, New York)
| N | Struktura | |||||||||||||||||||
| Con A | DSA | E4-PHA (20oC) | E4-PHA (4 oC) | L4-PHA | ||||||||||||||||
| 1 |
| |||||||||||||||||||
| +a | - | - | - | - | ||||||||||||||||
| 2 |
| |||||||||||||||||||
| + | - | - | r | - | ||||||||||||||||
| 3 |
| |||||||||||||||||||
| + | - | - | r | - | ||||||||||||||||
| 4 |
| |||||||||||||||||||
| + | +b | - | - | - | ||||||||||||||||
| 5 |
| |||||||||||||||||||
| + | + | - | r | - | ||||||||||||||||
| 6 |
| |||||||||||||||||||
| - | - | r | r | - | ||||||||||||||||
| 7 |
| |||||||||||||||||||
| - | r | - | r | - | ||||||||||||||||
| 8 |
| |||||||||||||||||||
| - | r | r | r | - | ||||||||||||||||
| 9 |
| |||||||||||||||||||
| - | + | - | r | - | ||||||||||||||||
| 10 |
| |||||||||||||||||||
| - | + | r | r | - | ||||||||||||||||
| 11 |
| |||||||||||||||||||
| - | + | - | - | r | ||||||||||||||||
| 12 |
| |||||||||||||||||||
| - | + | - | - | r | ||||||||||||||||
-metyloglukopiranozą w TB-NaN3b- wymywane 1% oligomerem N-acetyloglukozoaminy w TB-NaN3
Tabela 6. (Glycobiology 1993 s. 249, edy. Minoru Fokuda i Akira Kobata, Oxford University Press, New York)
| Oligosacharyd | |||||||||||||||||||||||||||||
| Lektyna | Gęstość wiązania | Hapten | |||||||||||||||||||||||||||
| |||||||||||||||||||||||||||||
| Conavalia ensiformis (Con A) | 15 mg/ml | -metyloglukoza (10 mM) | |||||||||||||||||||||||||||
| |||||||||||||||||||||||||||||
| Conavalia ensiformis (Con A) | 15 mg/ml | -metylomannoza (100mM) | |||||||||||||||||||||||||||
| |||||||||||||||||||||||||||||
| Conavalia ensiformis (Con A) | 15 mg/ml | -metylomannoza (100mM) | |||||||||||||||||||||||||||
| |||||||||||||||||||||||||||||
| Lens culinarisPisum sativum (lektyna z grochu) | 15 mg/ml | -metylomannoza (100mM) | |||||||||||||||||||||||||||
| |||||||||||||||||||||||||||||
| Lens culinarisPisum sativum (lektyna z grochu) | 15 mg/ml | -metylomannoza (100mM) | |||||||||||||||||||||||||||
| |||||||||||||||||||||||||||||
| Phaseolus volgaris(L4PHA) | 10 mg/ml | GalNAc (0.4 M) | |||||||||||||||||||||||||||
| |||||||||||||||||||||||||||||
| Phaseolus volgaris(L4PHA) | 10 mg/ml | GalNAc (0.4 M) | |||||||||||||||||||||||||||
| |||||||||||||||||||||||||||||
| Phaseolus volgaris(L4PHA) | 10 mg/ml | GalNAc (0.4 M) | |||||||||||||||||||||||||||
| |||||||||||||||||||||||||||||
| Phaseolus volgaris(L4PHA) | 8 mg/ml | mieszanina N,N"-di-N-acetylochitobiozy i N,N",N""-tri-N-acetylkochitotriozy | |||||||||||||||||||||||||||
| |||||||||||||||||||||||||||||
| Lycopersicon esculentum (lektyna z pomidorów) | 8 mg/ml | mieszanina N,N"-di-N-acetylochitobiozy i N,N",N""-tri-N-acetylkochitotriozy | |||||||||||||||||||||||||||
| |||||||||||||||||||||||||||||
| Lycopersicon esculentum (lektyna z pomidorów) | 15 mg/ml | mieszanina N,N"-di-N-acetylochitobiozy i N,N",N""-tri-N-acetylkochitotriozy | |||||||||||||||||||||||||||
n = 0,1 i >2 | |||||||||||||||||||||||||||||
| Datura stramonium(DSA) | 15 mg/ml | mieszanina N,N"-di-N-acetylochitobiozy i N,N",N""-tri-N-acetylkochitotriozy | |||||||||||||||||||||||||||
| |||||||||||||||||||||||||||||
| Maackia amurensis (MAL lub MAM) | 10 mg/ml | Laktoza (100 mM) | |||||||||||||||||||||||||||
| |||||||||||||||||||||||||||||
| Sambucus nigra aglutynina (SNA) | 10 mg/ml | Laktoza (100 mM) | |||||||||||||||||||||||||||
| |||||||||||||||||||||||||||||
| Griffonia simplicifolia (I-B4; GS-I-B4) | 15 mg/ml | Rafinoza (100 mM) | |||||||||||||||||||||||||||
| |||||||||||||||||||||||||||||
| Helix pomatia aglutynina (HPA) | 5 mg/ml | GalNAc (10 mM) | |||||||||||||||||||||||||||
| |||||||||||||||||||||||||||||
| Ricinus communis aglutynina (RCA-I) | 20 mg/ml | Laktoza (100 mM) | |||||||||||||||||||||||||||
| |||||||||||||||||||||||||||||
| Wisteria floribunda aglutynina (WFA) | 10 mg/ml | GalNAc (50 mM) | |||||||||||||||||||||||||||
| |||||||||||||||||||||||||||||
| Ulex europaesus I (UEA-I) | 10 mg/ml | Fuc (100 mM) | |||||||||||||||||||||||||||
- i -D-glikozydowych w procesie utleniania za pomocą CrO3
w środowisku kwasu octowego. Ponieważ wiązanie posiada dwa atomy tlenu
umieszczone blisko siebie (pochodzące z pierścienia i wiązania glikozydowego)
jest bardziej prawdopodobne, że ulegnie szybciej utlenieniu niż wiązanie
. Dalsza analiza polega porównaniu składu sacharydu przed i po procesie
utleniania za pomocą GC/MS. Obecność nieprzereagowanych cukrów sugeruje
występowanie wiązania -glikozydowego (Hoffman, Lidberg, Svensson 1972).
| ConA- | lektyna z kanawalii mieczoksztaltnej (Canavalia ensiformis) |
| D- | deuter |
| Da- | dalton |
| Fuc- | fukoza |
| Gal- | galaktoza |
| GalNAc- | N-acetylogalaktozoamina |
| Glc- | glukoza |
| GlcNAc- | N-acetyloglukozoamina |
| HPLC- | High Performance Liquid Chromatography |
| HPAEC- | High Performance Anion Exchanege Chromatography |
| IR- | promieniowanie podczerwone |
| Man- | mannoza |
| NeuAc- | kwas N-acetyloneuraminowy |
| SDS- | sól sodowa kwasu dodesylosulfonowego |
| Xyl- | ksyloza |