
|
34 35
36
37 |
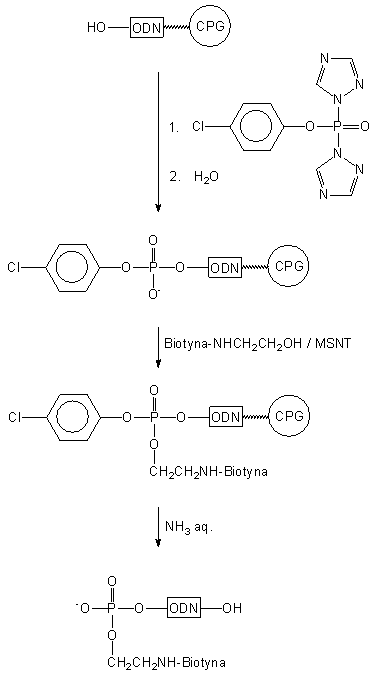 |
Wg podobnej koncepcji do fosforylacji oligomeru wykorzystano aktywne
pochodne fosforu trójwartościowego: PCl3 lub salicylochlorofosfinę
(Schemat 5)46. W obu przypadkach po pierwszym etapie, tj. po
przyłączeniu fosforynu do oligomeru, aktywne centrum fosforowe poddawano
hydrolizie do H-fosfonianu, który w kolejnej reakcji z N-1-trifluoroacetyloetanoloaminą
wobec chlorku piwaloilu ulegał estryfikacji. Po utlenieniu i odblokowaniu
otrzymywano oligonukleotyd z grupą aminoalkilową przyłączoną poprzez resztę
fosforanową na końcu 5'.
|
34 38
39
40 |
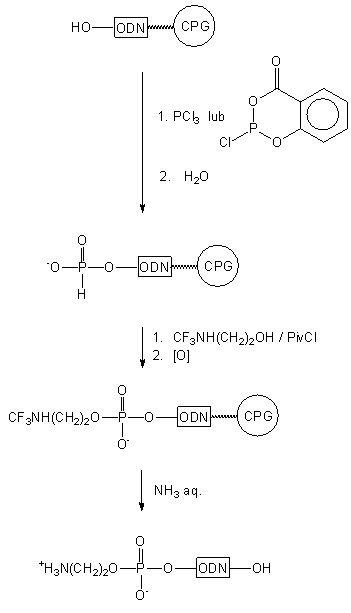 |
Analogiczny produkt otrzymano w podobnym cyklu reakcji (Schemat 6),
jednakże łącznik aminoalkilowy wprowadzano na etapie oksydatywnej kondensacji
5'-(metoksy-H-fosfonianu) oligomeru z diaminoetanem46.
|
34 41
42
43 |
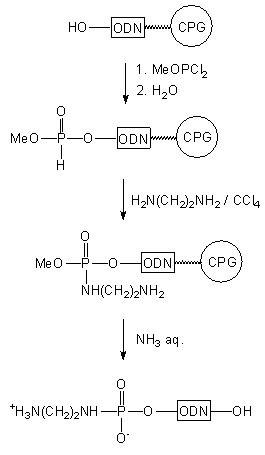 |
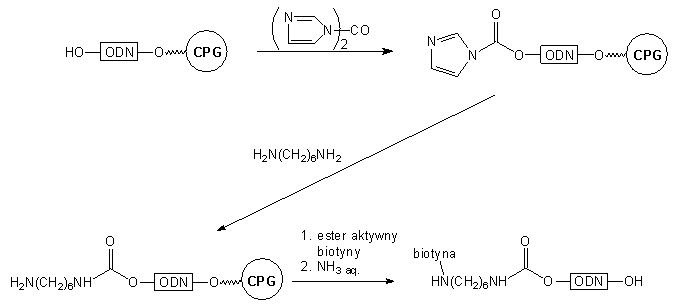
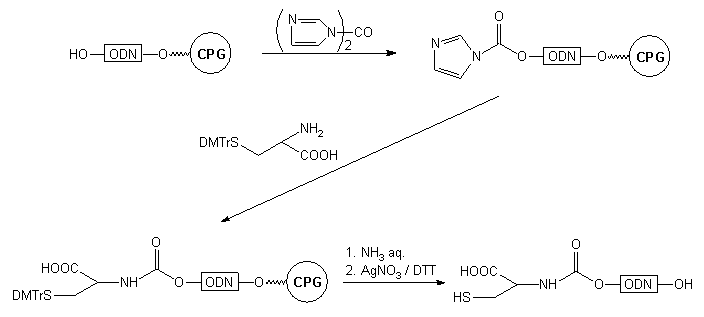
Odmiennym typem reagentów generujących aktywny układ na końcu 5' są
karbodiazolidy47-49. Aktywacja oligonukleotydu 34
karbonylodiimidazolem prowadzi do pochodnej typu 44, nieaktywnej
wobec alkoholi, natomiast łatwo reagującej z aminami50. W wyniku
reakcji z 1,6-diaminoheksanem uzyskano karbaminian typu 45.
Po przyłączeniu biotyny i odblokowaniu roztworem amoniaku (reszta karbaminianowa
jest trwała w tych warunkach49) otrzymano gotową sondę oligonukleotydową
(Schemat 7).
|
34 |
 |
44 |
| 45 | 46 |
Schemat 7
Podobnie przebiegły reakcje prowadzące do 5'-SH oligomeru47
(Schemat 8). W tym przypadku pochodną karbonyloimidazolową typu 44
potraktowano S-dimetoksytrytylocysteiną. Produktem tej reakcji był oligomer
47
z dowiązaną na końcu 5' cysteiną (poprzez ugrupowanie karbaminianowe).
Resztę tiolową cysteiny wykorzystano po odblokowaniu do dowiązania grupy
pirenylowej. Oprócz grupy -SH oligomer 48 niesie na końcu
5' również grupę karboksylową, potencjalne miejsce selektywnego przyłączania
innego typu grupy reporterowej.
|
34 |
 |
44 |
| 47 | 48 |
Schemat 8
Również podobny system funkcjonalizacji zastosowano do otrzymania sondy oligonukleotydowej znakowanej wieloma grupami reporterowymi48. Na pierwszym etapie sprzęgano 1,6-diaminoheksan z oligomerem wobec karbonylodiimidazolu. Stosując następnie dwukrotną kondensację z Na -Ne -di-Fmoc-lizyną wobec BOP przyłączono do grupy -NH2 cztery reszty lizyny. Ochronne grupy Fmoc usuwane były selektywnie działaniem DBU, a do uwolnionych w ostatnim etapie grup aminowych przyłączane były cząsteczki biotyny, prowadząc do związku typu 49. Po odcięciu od podłoża i odblokowaniu w standardowych warunkach izolowano tetrabiotynylowaną sondę oligonukleotydową techniką HPLC lub PAGE.
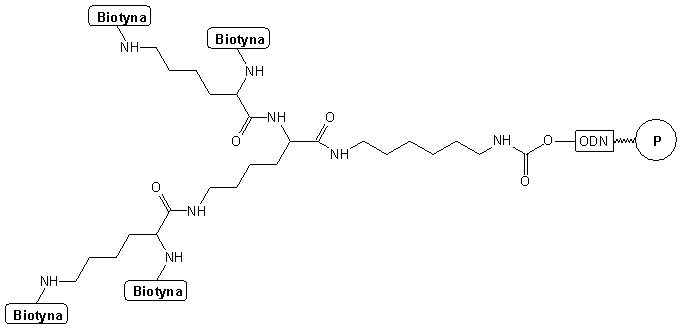
49
50
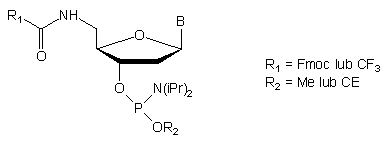
Dla realizacji takiego podejścia zaproponowano użycie związków typu 50 jako końcowej modyfikowanej jednostki nukleotydowej51,52. Labilne w warunkach zasadowych grupy chroniące resztę aminową usuwane były w trakcie końcowego odblokowania roztworem amoniaku. Po syntezie oligomeru, kondensacji z amidofosforynem typu 50 i odblokowaniu, reszta aminowa w pozycji 5' uwolnionego oligomeru wykorzystana była do wprowadzania fluorescencyjnych grup reporterowych.
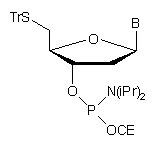
51
W celu uzyskania oligonukleotydów z grupą -SH na końcu 5' zaproponowano analogiczne do opisanych powyżej syntony typu 5153. Lipofilowa grupa trytylowa na końcu 5' oligonukleotydów otrzymanych z wykorzystaniem z tego typu syntonów umożliwiała łatwe oczyszczanie produktu za pomocą RP-HPLC*. Grupę trytylową usuwano jonami srebra lub rtęci.
Przedstawione powyżej metody wydają się mało atrakcyjne z dwóch powodów: (i) konieczność syntezy modyfikowanych nukleozydów i ich amidofosforynów; (ii) bezpośrednie przyłączenie grupy funkcyjnej do węgla 5' reszty cukrowej może powodować powstawanie znacznej zawady przestrzennej po przyłączeniu grupy reporterowej, co może mieć negatywny wpływ na proces hybrydyzacji i detekcji.
Ze względu na znaczą liczbę opisanych metod syntezy i zastosowań syntonów nienukleotydowych zestawiono je w tabelach i opatrzono komentarzem podkreślającym istotne właściwości chemiczne i ich konsekwencje.
Tabela 3. Amidofosforyny wprowadzające grupy funkcyjne
| Nr | Związek | Komentarz | Lit. |
| 52 | 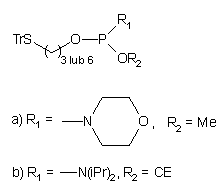 |
|
54, 55 |
| 53 | 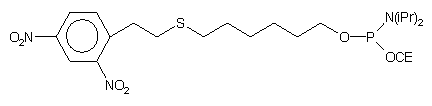 |
NH3 aq. stęż./0,1 M DTT |
56 |
| 54 | 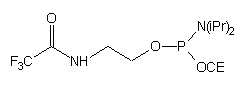 |
|
57 |
| 55 | 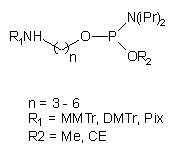 |
|
54, 58, 59 |
| 56 | 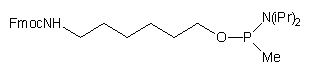 |
|
60 |
| 57 | 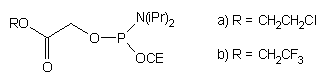 |
|
61 |
| 58 | 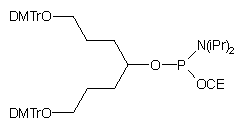 |
|
62 |
| 59 | 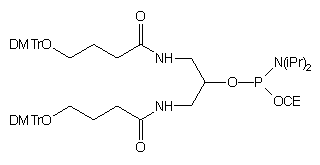 |
63 | |
| 60 | 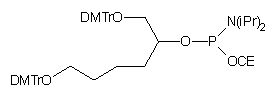 |
19 | |
| 61 | 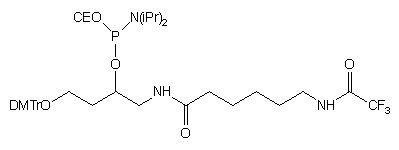 |
|
19 |
| 62 | 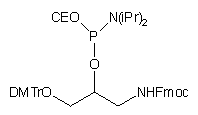 |
|
64 |
| 63 | 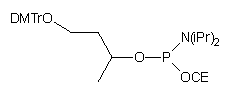 |
|
19 |
Spośród syntonów funkcjonalizujących chciałbym wyróżnić amidofosforyn
typu 55. Zawiera on względnie prosty szkielet węglowy (co
ułatwia jego przygotowanie) i po przyłączeniu do oligomeru umożliwia kolorymetryczny
pomiar wydajności funkcjonalizacji. Dodatkowo chemoselektywne usunięcie
protekcji grupy funkcyjnej pozwala na przyłączenie grupy reporterowej do
oligomeru związanego z podłożem, co znacznie upraszcza procedurę izolacji
finalnego produktu syntezy. Alternatywnie, odblokowanie oligomeru z zachowaniem
lipofilowej blokady dimetoksytrytylowej stwarza możliwość szczególnie skutecznego
oczyszczenia sfunkcjonalizowanego oligonukleotydu za pomocą RP-HPLC.
Tabela 4. Amidofosforyny wprowadzające grupy reporterowe
| Nr | Związek | Komentarz | Lit. |
| 64 | 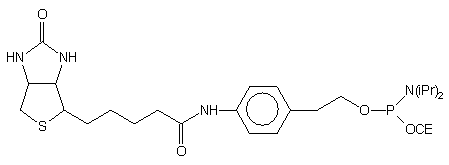 |
|
65, 66 |
| 65 | 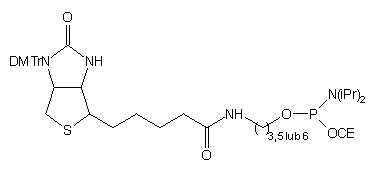 |
|
67, 68 |
| 66 | 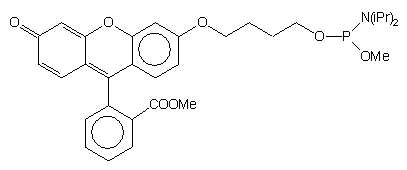 |
|
69 |
| 67 | 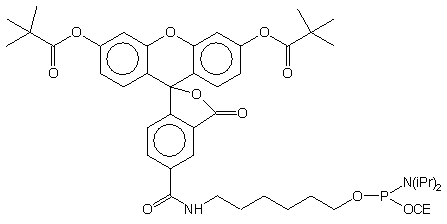 |
|
70 |
| 68 | 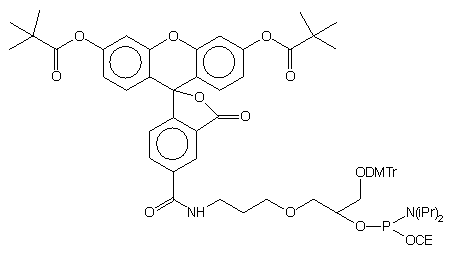 |
|
41 |
| 69 | 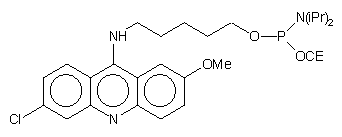 |
|
28, 71 |
| 70 | 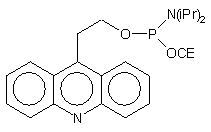 |
|
72 |
| 71 | 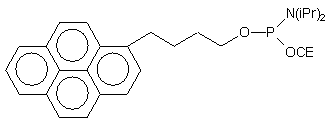 |
|
63, 73 |
| 72 | 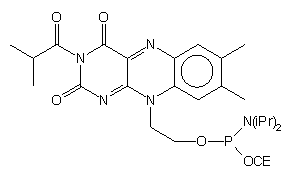 |
|
74 |
| 73 |  |
|
75 |
| 74 | 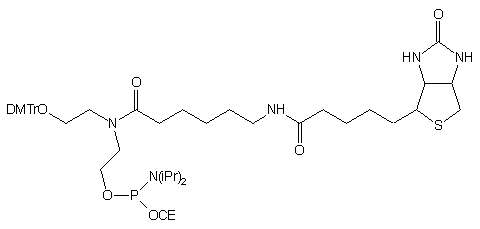 |
|
76 |
| 75 | 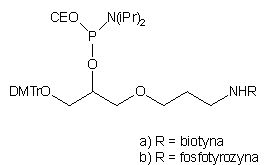 |
|
77 |
| 76 | 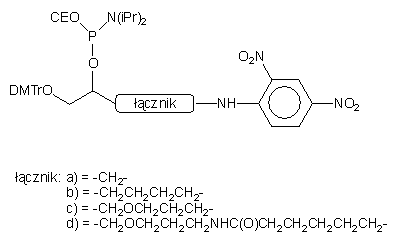 |
Rodzaj łącznika miał mały wpływ na poziom detekcji oligomeru.
|
79 |
Wykorzystanie syntonów 64 - 76 wydaje się
być najprostszą metodą wprowadzania grup reporterowych w pozycji 5' oligonukleotydów.
Należy jednak zwrócić uwagę, że często synteza tego typu związków jest
procesem wieloetapowym, a produkt końcowy uzyskiwany jest z niską wydajnością.
Ograniczone jest też spektrum możliwych do wykorzystania grup reporterowych,
gdyż muszą one być odporne na warunki kondensacji, utleniania i odblokowania
oligomeru. Ponadto wprowadzenie innego znacznika do oligonukleotydu wiąże
się z koniecznością syntezy nowego syntonu. W związku z tymi niedogodnościami
uważam, że korzystniejszym podejściem jest funkcjonalizacja oligomeru,
a następnie przyłączenie grupy reporterowej.
77 |
R1 = Me, CE, Npe
R2 = CE, Npe |
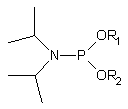
Jednymi z pierwszych reagentów zastosowanych z powodzeniem do fosforylacji oligonukleotydu związanego z podłożem stałym były N,N-diizopropyloamidofosforyny dialkilowe typu 7780. Produktem reakcji fosforynacji był 5'-dialkilofosforyn oligonukleotydu, utleniany następnie do fosforanu. Grupy alkilowe usuwano działaniem: tiofenolanu (R=Me), amoniaku (R=CE) lub DBU (R=Npe), uzyskując 5'-fosforylowany oligomer.
78
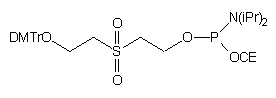
Przykładem innego czynnika fosforylującego, który prowadził do analogicznego 5'-fosforylowanego oligonukleotydu jest N,N-diizopropyloamidofosforyn 2-cyjanoetylowo - 2-[2'-(4,4'-dimetoksytrytyloksy)etylosulfonylo]etylowy (78)81. Obok "aktywnego" ugrupowania amidofosforynowego i ochronnej grupy 2-cyjanoetylowej zastosowano ugrupowanie 2-(2'-hydroksyetylosulfonylo)etylowe. To ostatnie zawierało grupę dimetoksytrytylową, co umożliwiało monitorowanie (test barwny) reakcji fosforynacji. Obydwie grupy fosforanoalkilowe [2-cyjanoetylową i 2-(2'-hydroksyetylosulfonylo)etylową] usuwano w reakcji b -eliminacji w środowisku zasadowym.
79
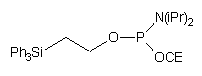
W innym podejściu, do fosforylacji oligomeru zastosowano N,N-diizopropyloamidofosforyn 2-cyjanoetylowo - 2-(trifenylosililo)etylowy (79)82. Po utlenieniu i odblokowaniu roztworem amoniaku, grupa 2-(trifenylosililo)etylowa wciąż pozostawała związana z resztą 5'-fosforanową. Dzięki temu możliwe było łatwe oczyszczenie (za pomocą RP-HPLC) pożądanego produktu od zanieczyszczeń nie niosących tego silnie lipofilowego ugrupowania. Grupę 2-(trifenylosililo)etylową usuwano następnie jonami fluorkowymi, uzyskując 5'-fosforan oligonukleotydu.
80
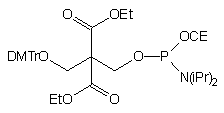
Kolejnym amidofosforynem stosowanym do fosforylacji oligonukleotydów jest związek 8083. Otrzymano go z 2,2-bis(hydroksymetylo)malonianu dietylowego poprzez dimetoksytrytylowanie i fosfitylację. Zastosowanie tego amidofosforynu umożliwia dwutorowe wykorzystanie właściwości grupy dimetoksytrytylowej: kontrolowanie wydajności reakcji fosforylacji dzięki uwalnianiu barwnego kationu dimetoksytrytylowego (metoda "trityl off") lub wydajne oczyszczanie oligomeru dzięki lipofilowym właściwościom tej grupy (metoda "trityl on"). To drugie podejście jest możliwe dzięki nieoczekiwanej trwałości estru 2,2-bis(etoksykarbonylo)-3-(4,4'-dimetoksytrytyloksy)propylowego w stężonym roztworze amoniaku. Natomiast po usunięciu grupy dimetoksytrytylowej, ugrupowanie to staje się nietrwałe w warunkach zasadowych i traktowanie oligomeru wodnym roztworem amoniaku lub aminy pozwala na uzyskanie 5'-fosforanu oligonukleotydu (Schemat 9).
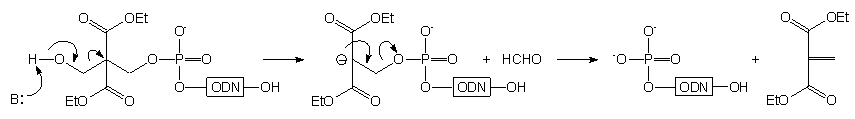
Schemat 9
Opisane powyżej podejścia z wykorzystaniem amidofosforynów prowadziły do 5'-fosforanów oligonukleotydów. Lepsze możliwości przyłączania innych reagentów modyfikujących czy grup reporterowych na końcu 5' wydaje się stwarzać ugrupowanie wodorofosforynowe. Może być też ono traktowane jako modyfikacja oligomerów służąca np. do badań aktywności substratowej w reakcjach enzymatycznych.
Najprostsze reagenty służące do otrzymania 5'-wodorofosforynów oligonukleotydów to opisane wcześniej (*) PCl3 i salicylochlorofosfina46. Ze względu na dużą reaktywność stwarzają one jednak zagrożenie modyfikacji zasad azotowych łańcucha oligonukleotydowego.
Bardziej odpowiednia dla selektywnego modyfikowania 5'-końcowej reszty
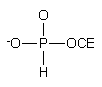
hydroksylowej oligonukleotydów jest metoda z użyciem monoestrów H-fosfonianowych
8184
lub 8285, zawierających podatne na b
-eliminację reszty alkilowe.
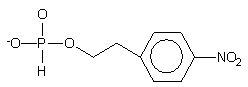 |
 |
|
|
|
Związek 81 [H-fosfonian 2-(p-nitrofenylo)etylowy] otrzymano przeprowadzając PCl3 w tris(1,2,4-triazoilo)fosforyn, który poddawano reakcji z 2-(p-nitrofenylo)etanolem. Po hydrolizie powstałego p-nitrofenylofosforoditriazolidu, produkt 81 oczyszczano na żelu krzemionkowym. H-Fosfonian 2-cyjanoetylowy 82 otrzymano przez hydrolizę dichlorofosforynu 2-cyjanoetylowego i stosowano bez oczyszczania. Związki powyższe umożliwiają fosfonylację oligonukleotydów w reakcji standardowej kondensacji z chlorkiem piwaloilu. b-Eliminacja reszty 2-(p-nitrofenylo)etylowej następuje w wyniku działania DBU, reszta 2-cyjanoetylowa zaś ulega b -eliminacji w trakcie końcowego działania amoniakiem. W obu przypadkach uzyskuje się 5'-wodorofosforyny oligonukleotydów.
W powyższych podejściach zastrzeżenia może budzić brak zadowalającej procedury uzyskiwania czystych monoestrów. Opisana została jednak inna, prosta metoda otrzymywania wodorofosforynów monoalkilowych poprzez transestryfikację H-fosfonianu difenylowego odpowiednim alkoholem. Powstający na pierwszym etapie H-fosfonian dialkilowy hydrolizuje się do monoestru, który wydziela się jako produkt krystaliczny86.
Marsters i wsp.87 opublikowali metodę bezpośredniej fosfonylacji oligonukleotydu kwasem fosforawym z użyciem chlorku piwaloilu jako czynnika aktywującego. Przy proponowanym sposobie aktywacji kwasu fosforawego i stosowanych nadmiarach reagentów produktem głównym (~90%) reakcji był symetryczny H-fosfonian 5'-5' dioligonukleotydowy (83). Zwiększenie ilości kwasu fosforawego względem chlorku piwaloilu w roztworze fosfonylującym poprawiło wydajność 5'-H-fosfonianu oligonukleotydu (84), jednakże 5'-5' symetryczny dimer był nadal jednym z głównych (~40 %) produktów reakcji (Schemat 10). Przy dalszym wzroście nadmiaru H3PO3 fosfonylacja zachodziła ze zdecydowanie mniejszą wydajnością.
W zamierzeniach autorów metody, jej głównym walorem miało być stosowanie
prostych i tanich reagentów, a także prostej chemii prowadzącej bezpośrednio
do pożądanego produktu. Otrzymane wyniki negatywnie zweryfikowały te oczekiwania.
Według mojej oceny podejście to nadal zasługuje na uwagę, jednakże wymaga
dopracowania metodycznego. Uważam, że warunkiem koniecznym kontroli procesu
fosfonylacji w proponowanym systemie jest identyfikacja czynnika fosfonylującego.
Bez większego ryzyka można postulować, że układ produktów opisany przez
Mastersa i wsp. wiąże się nie z samą reakcją fosfonylacji, lecz ze sposobem
aktywacji kwasu fosforawego chlorkiem piwaloilu i kontrola tej ostatniej
reakcji może znacznie poprawić chemoselektywność procesu.
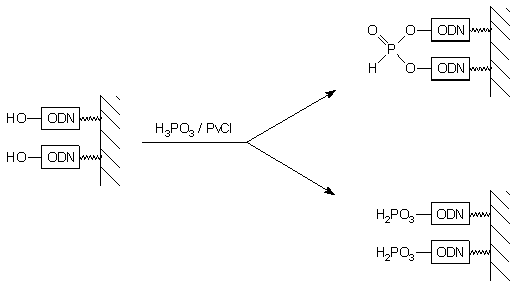 |
83
84 |
Schemat 10
